

Erika Update #1: Getting projects started is hard
“Nothing works/makes sense. Going home.”

Choose a good environment
It was obvious early on in my PhD that the automation part of what I wanted to do would work well. What was more uncertain was which biological system I would apply it to. I spent the first few years of graduate school pitching project after project and never getting traction on any of them. There were a lot of things working against me, including that I was part of a brand new lab (Kevin Esvelt’s lab), Kevin was very hands-off, there were no postdocs, I was trying to work with areas of biology that Kevin hadn’t personally worked with before, and I had very little experience with wet lab biology myself.
In late Fall 2017, I pitched the qtRNA project. Unlike other projects, it happened to fit nicely into the research portfolio of Ahmed Badran, who had just started his own mini-group across the street at the Broad Institute. We decided we’d collaborate, and he’d give me some hands-on mentorship to get the project going. I started wet lab work when I got back from break in January 2018. The first milestone in a biomolecule engineering project is to create a reliable way to measure whether or not biomolecules you engineer work. I used a luciferase reporter for that, which was quite similar to what had been done in the literature, so I expected it to work.
In early February, I tested the luciferase reporter together with some qtRNAs the literature said were functional. Contrary to my expectations, there was absolutely no signal. Zip. I spent one dark night convinced that all of synthetic biology was a lie and that I should drop out of grad school and go into high-frequency trading, and then the next morning woke up, checked my plasmids, and realized I had cloned the anticodon into the tRNA rather than the reverse complement of the anticodon (I had gotten it backwards and upside-down), and so naturally it didn’t work. I recloned correctly and a week later it worked just fine and I didn’t drop out of grad school. I had a functional reporter!
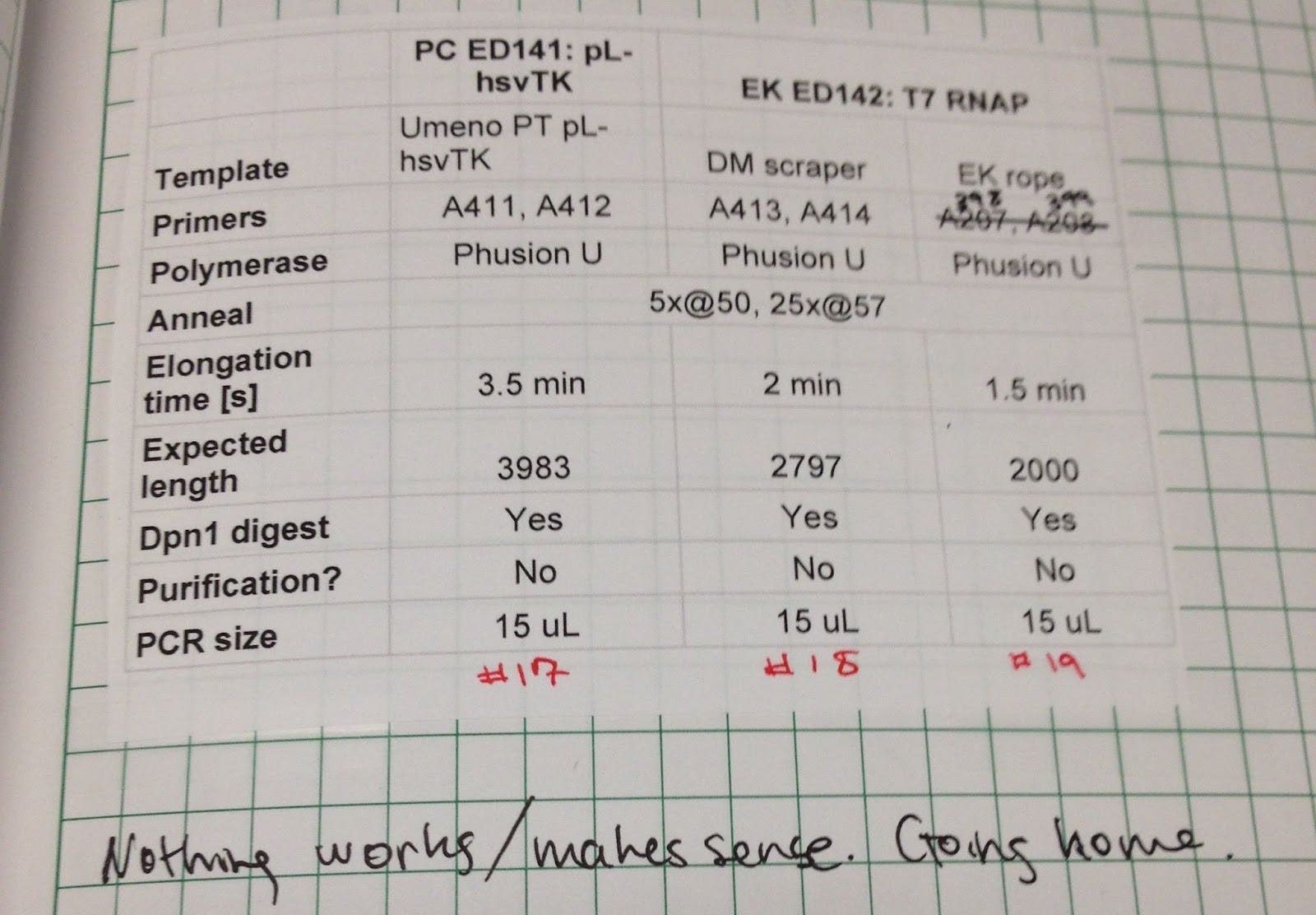
It’s worth noting that getting this far required both a lot of perseverance on my part and a favorable environment with good technical support.
Erika Update #1
As a quick recap, this post is part of the “Erika Updates” series, where I post informal research updates I wrote in grad school accompanied by blog posts with commentary. I’m revisiting these write ups as a jumping off point for reflecting on the PhD experience, the current state of research science, and the ways we share and communicate technical content to peers and the public.
Here’s Erika Update #1: 2018 2 20 - luxAB frameshift reporter
This is the first Erika Update I wrote about this project, and corresponded to the first tiny solid step forward. I sent it out by email four years ago today!
Where’d it end up?
It wasn’t until later that I figured out an improved way to run this experiment and re-measure all the samples. So this particular batch of data was never published, but its successor became quite central data to both Paper #1 and Paper #2.
And to orient you
I think pretty much everyone can get something out of reading these blog posts + updates. Here’s some additional notes customized for you, depending on your interests + background:
You’re a non-scientist: I’m trying to engineer a type of biomolecule called a ‘tRNA.’ In this document, I show that I can measure how well a tRNA is working by using a luciferase, which is the gene that makes fireflies glow. I engineered a sensor so that the better the tRNA works the more my samples glow! In the graph, the fact that the bar on the right is higher in each colored pair means that the tRNAs I’m testing are working. That’s the main result.
You’re a student doing a PhD/undergrad research/etc: The nomenclature I use for primers is “ED####”. I restarted my numbering for this project because I was working in a new lab, and primer ED0001 was one of the primers used to clone a frameshift into residue 357 of my luciferase gene. I use ED0001 to this day - literally the same tube. Turns out making reporters and having a way to quantify whether anything you do is working is very essential and sticks with you throughout a project.
You have ideas for reforming publishing: This is a good example of an Erika-Update that I think really is too small to be appropriate for publishing alone: it is only about halfway through replicating the first key points in the literature. I see this Erika-Update as an argument against infinitely micro publishing, but perhaps in favor of internal unpublished writeups.
You’re a fellow PACE nerd: The literature mostly inserted four-base codons into GFP (for a fluorescence readout), or antibiotic genes like chloramphenicol resistance (to create an antibiotic resistance selection). The PACE community more commonly uses luciferase as the reporter of choice for a few reasons 1) the standard PACE strain David Liu’s lab uses (S2060s, available on Addgene) has additional luciferase genes encoded on the F plasmid that produce the substrate endogenously, making it very easy to do luciferase assays. You don’t need to add substrate or anything, just use that strain of E coli. 2) Luciferases are more sensitive on the low-end, so they’re ‘better’ than GFP at the beginning of an evolution project, where you want to compare amongst low-activity variants. 3) Ahmed Badran told me that luciferases mature at a kinetic rate that is more similar to pIII than GFP, making luciferase results ‘track’ later phage enrichment results more closely than GFP results. I haven’t tested this, but that’s the lore.
You’re a fellow genetic code expansion nerd: I selected two different sequence contexts for the frameshift right off the bat, but sequence context didn’t end up being a line of inquiry I delved into very deeply.
Vocabulary highlight: “Molecular Cloning”
‘Molecular cloning’ is the process of creating custom pieces of DNA that encode new instructions for the cell.
Cloning in biological engineering is the equivalent of writing a program in computer science. In both cases, you have an idea for a new gene/program you want to test. You need to encode that gene/program in a format your cell/computer can understand: DNA/ 0’s & 1’s.
‘Cloning’ is a very confusing name because it suggests you’re making an army of identical little green aliens, which is not required. Historically, the name comes from the same idea on the molecular level. Fifty years ago when people first started doing biological engineering, you couldn’t just synthesize any DNA you wanted. Instead, you would begin with pieces of DNA that already existed, copy them (“molecular cloning”!), and stitch the copies together in new ways. Although technology has improved, to this day life science researchers spend an enormous fraction of their time cloning.
Want more?
I had a lot of fun with this project during my PhD, and I hope that this publishing experiment will be a fun read! If you want to follow along, you can get updates by signing up through substack below. I’ll also post on linkedin and twitter. If you’re curious about more background on the project, Quanta/Wired wrote an excellent article on it, and I also talked a lot about this project as a guest on the molpigs podcast.
If you have ideas for what I should cover in the blog post, suggestions for vocabulary to define, questions about the science, or other comments, please do reach out by twitter DM - I’d love to hear from you!